
Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy

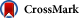
Open access funded by Dutch Universities
 Article Info
Article Info
Fig. 1
Functional characterization of the p.Arg223Gly mutant in Nav1.6. Neuronal ND7/23 cells were transfected with wildtype or mutant Nav1.6 cDNA. (A) Cells were grown overnight at the normal temperature of 37 °C or reduced temperature of 30 °C and recorded at room temperature. Superimposed traces recorded in response to the activation stimulation protocol (100 msec duration pulses) are shown. (B) Peak currents were normalized for cell capacitance and averaged to obtain the expressed current-density for cells expressing mNav1.6r-WT (light grey, n = 14 at 37 °C, 9 at 30 °C) or mNav1.6r-Arg223Gly (dark grey, n = 15 at 37 °C, 12 at 30 °C) grown at 37 °C or 30 °C overnight. (C) Activation G–V curves were normalized and averaged as described in ‘Methods’. (D) Responses to the fast-inactivation protocol are analysed to obtain the voltage-dependence of fast-inactivation as described in ‘Methods’. Error bars are standard error of the mean (SEM). The averages of the normalized G–V curves for activation for Arg223Gly and WT channels grown at 37 °C or 30 °C were not significantly different (C). See Supplement for data.
Fig. 2
Enhanced ramp activity of Nav1.6 resulting from the p.Arg223Gly mutation. Data traces recorded in response to a slow ramp stimulus of −120 mV to +20 mV over 600 ms are filtered to 200 Hz and normalized to peak I–V current. The normalized ramp traces are averaged together for cells expressing Nav1.6-WT (black line, n = 5) or Nav1.6-Arg223Gly channels (grey line, n = 11). Selected points are plotted as mean ± SEM to indicate the variance of the averages. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3
The p.Arg223Gly mutation reduces the stability of the Nav1.6 channel protein. HEK293 cells were transfected with mutant or wildtype Nav1.6 cDNA and cultured for 24 h at 37 degrees. Aliquots of cell extracts containing 30 μg protein were subjected to electrophoresis on 4–15% gradient gels and transferred to a nitrocellulose filter as described in ‘Methods’. Nav1.6 was detected with a rabbit polyclonal antibody (Alomone). As a control for loading and transfer efficiency, the filter was washed and the endogenous FIG4 protein was immunostained with a mouse monoclonal antibody (NeuroMab). Mu, mutant; Wt, wildtype; Un, untransfected; o, gel origin.
Highlights
- •Second functional characterization of SCN8A mutation in epileptic encephalopathy.
- •Channel functioning was investigated in neuronal cells.
- •Protein stability was investigated after correction for transcript abundance.
- •Functional effects of this mutation only partially overlapping with previously described mutation.
- •Raises discussion about functional mechanism of sodium channels in epileptic encephalopathy.
Summary
Objective
Recently, de novo SCN8A missense mutations have been identified as a rare dominant cause of epileptic encephalopathies (EIEE13). Functional studies on the first described case demonstrated gain-of-function effects of the mutation. We describe a novel de novo mutation of SCN8A in a patient with epileptic encephalopathy, and functional characterization of the mutant protein.
Design
Whole exome sequencing was used to discover the variant. We generated a mutant cDNA, transfected HEK293 cells, and performed Western blotting to assess protein stability. To study channel functional properties, patch-clamp experiments were carried out in transfected neuronal ND7/23 cells.
Results
The proband exhibited seizure onset at 6 months of age, diffuse brain atrophy, and more profound developmental impairment than the original case. The mutation p.Arg233Gly in the voltage sensing transmembrane segment D1S4 was present in the proband and absent in both parents. This mutation results in a temperature-sensitive reduction in protein expression as well as reduced sodium current amplitude and density and a relative increased response to a slow ramp stimulus, though this did not result in an absolute increased current at physiological temperatures.
Conclusion
The new de novo SCN8A mutation is clearly deleterious, resulting in an unstable protein with reduced channel activity. This differs from the gain-of-function attributes of the first SCN8A mutation in epileptic encephalopathy, pointing to heterogeneity of mechanisms. Since Nav1.6 is expressed in both excitatory and inhibitory neurons, a differential effect of a loss-of-function of Nav1.6 Arg223Gly on inhibitory interneurons may underlie the epilepsy phenotype in this patient.
